Integrating snRNA-seq with spatial transcriptomics for brain mapping
Why one technology is not enough for brain tissue
The brain is the most architecturally complex organ in the body. A single cortical region can contain dozens of cell types -- excitatory neurons, inhibitory interneurons, astrocytes, oligodendrocytes, microglia, vascular cells -- organized in layers and circuits that drive function. Disease processes disrupt these patterns in ways that require both spatial context and molecular resolution to understand.
Researchers who rely on spatial transcriptomics alone can map where cells sit in the tissue, but current spatial platforms use gene panels that cover only a fraction of the transcriptome. Researchers who rely on snRNA-seq alone get whole-transcriptome depth for every nucleus, but lose all spatial context the moment the tissue is dissociated. The most informative brain studies now combine both: spatial data provides the tissue map, snRNA-seq provides the cell-type annotations that make that map interpretable.
TL;DR -- paired spatial + snRNA-seq for brain FFPE
- Spatial transcriptomics maps cell locations but uses limited gene panels -- snRNA-seq adds whole-transcriptome resolution
- snRNA-seq cell-type clusters become the reference atlas that annotates spatial datasets
- Adjacent sections from the same FFPE block feed both arms of the experiment
- The Singulator 200+ produces platform-agnostic nuclei validated for 10x Flex and PERFF-seq, with snRNA-seq data serving as companion to Xenium spatial analysis
- Consistent nuclei quality from the S200+ means the snRNA-seq arm matches the spatial arm -- no prep-induced artifacts
Building a paired spatial + snRNA-seq brain study
Five practical considerations for designing multi-omic brain tissue experiments using FFPE sections and automated nuclei extraction.
Why brain needs both approaches Understand why brain tissue demands paired analysis
Brain architecture is not decorative. Where a neuron sits -- which cortical layer, which nucleus, which circuit -- determines what it does. An excitatory neuron in layer 2/3 of the visual cortex has a fundamentally different role than one in layer 5, even though both are glutamatergic. This means spatial context is not a nice-to-have; it is part of the biology.
But cell identity in the brain goes beyond location. Transcriptomic profiling has revealed cell-type diversity that histology alone never captured. The Allen Brain Cell Atlas, for instance, has catalogued hundreds of transcriptionally distinct cell types across the mouse brain, many of which are indistinguishable by morphology or marker staining. You cannot see these subtypes in a spatial experiment unless you already know what to look for.
The convergence happening now
Leading neuroscience groups have recognized that neither technology alone is sufficient. Spatial transcriptomics gives you the architecture. snRNA-seq gives you the cell-type resolution to populate that architecture with meaningful annotations. The Dana Pe'er lab at Memorial Sloan Kettering used this paired approach with Xenium spatial and Singulator 200+ nuclei to study mouse brain melanoma metastasis -- using spatial data to see where immune cells infiltrated the brain parenchyma, and single-nuclei data to characterize what those infiltrating populations were actually doing at the transcriptomic level.
Think of spatial transcriptomics as the map and snRNA-seq as the legend. A map without a legend tells you where things are but not what they are. A legend without a map tells you what exists but not where. Brain tissue studies published in the highest-impact journals increasingly include both.
Researchers sometimes assume that as spatial gene panels expand, snRNA-seq will become redundant for brain studies. Current spatial panels cover hundreds to thousands of genes -- whole-transcriptome snRNA-seq captures 20,000+. For rare cell-type discovery and novel transcript identification, the depth gap remains substantial.
What spatial tells you (and misses) Recognize what spatial platforms capture and what they miss
Spatial transcriptomics platforms like 10x Xenium and Visium analyze tissue sections in situ -- no dissociation required. The tissue stays intact on the slide, and the platform measures gene expression at specific locations. For brain tissue, this preserves the laminar organization, regional boundaries, and cell-cell spatial relationships that dissociation destroys.
What spatial does well
- Tissue architecture: Maps gene expression back to specific locations within cortical layers, white matter tracts, hippocampal subfields
- Cell neighborhoods: Identifies which cell types sit adjacent to each other -- neuron-glia interactions, immune infiltration patterns
- Pathology context: Overlays molecular data onto histological features visible in the same section
What spatial misses
Current spatial platforms use pre-designed gene panels. Even the largest panels cover a fraction of the transcriptome. If a cell type is defined by expression of genes outside the panel, or if you are looking for novel transcripts not included in the probe set, spatial alone will not find them. Cell-type assignment from spatial data also depends on reference atlases built from -- you guessed it -- single-cell or single-nuclei sequencing.
Xenium panels currently cover 100-5,000 genes depending on the panel configuration. Whole-transcriptome snRNA-seq routinely detects 15,000-20,000 genes per sample. For brain regions with high cell-type diversity (hippocampus, cortex), the unbiased discovery power of snRNA-seq often identifies populations that pre-designed panels miss.
What snRNA-seq adds Add whole-transcriptome depth with snRNA-seq
Single-nuclei RNA sequencing dissociates the tissue, captures individual nuclei, and sequences their entire transcriptome. For FFPE brain tissue, this means using probe-based sequencing chemistry -- 10x Genomics Flex is the most widely adopted platform for this -- because formalin-fixed RNA is too fragmented for standard poly-A capture methods. Flex probe pairs have a compact 50-nucleotide footprint, bypassing the need for intact poly-A tails, making it well-suited for degraded FFPE material.
The cell-type discovery advantage
Brain tissue processed through snRNA-seq generates data for unsupervised clustering -- the algorithm groups nuclei by transcriptomic similarity without any prior assumptions about what cell types should be present. This is how researchers discover new cell subtypes, identify disease-associated populations, and build the reference atlases that spatial experiments depend on for cell-type annotation.
The nuclei quality bottleneck
All of this depends on the quality of nuclei going into the sequencing workflow. Debris-laden nuclei suspensions produce high ambient RNA backgrounds, reduce the fraction of reads mapping to cells, and bias cell-type representation toward populations that survived the extraction process. For brain FFPE tissue specifically, myelin debris and lipid contamination from the high-fat neural tissue create additional challenges that make the nuclei extraction step the single biggest determinant of data quality.
For paired spatial + snRNA-seq studies, inconsistent nuclei quality between the snRNA-seq samples undermines the cross-modal comparison. If your snRNA-seq reference atlas is biased toward immune cells because manual trituration destroyed fragile neuronal nuclei, your spatial cell-type annotations will inherit that bias. The nuclei prep determines the quality of both datasets.
When planning snRNA-seq from FFPE brain tissue, check DV200 before committing your section. A DV200 above 50% is ideal for Flex chemistry. Between 30-50%, sequencing is possible but expect lower gene detection per nucleus. Below 30%, consider spatial-only approaches that do not require dissociation.
Design paired experiments Design paired experiments from the same FFPE block
The practical advantage of FFPE brain tissue for multi-omic studies is that a single block can yield multiple sections for different analyses. One section goes on a spatial transcriptomics slide. An adjacent section gets processed for snRNA-seq. Because the sections come from the same block -- separated by micrometers rather than millimeters -- the cellular composition is as closely matched as biologically possible.
Block allocation strategy
Talk to your neuropathologist or biobank coordinator before sectioning. Plan the cut order so that spatial sections and snRNA-seq sections are as close together as possible on the block face. A common approach: cut 5-10 micrometer sections for spatial, then immediately cut a 50-micrometer curl for snRNA-seq from the same face.
| Analysis arm | Section type | Processing |
|---|---|---|
| Spatial (Xenium/Visium) | 5-10 micrometer on slide | On-slide in situ, no dissociation |
| snRNA-seq (10x Flex) | 50 micrometer curl | GREEN then YELLOW NIC+ S200+ Only |
| PERFF-seq (rare cells) | 50 micrometer curl | GREEN then YELLOW NIC+ S200+ Only |
For longitudinal or cohort studies, plan the sectioning scheme across the entire study before cutting the first block. A 50-micrometer curl from the Singulator 200+ typically yields over 1 million nuclei -- far more than the 10,000-20,000 needed for a standard Flex capture. This means a single curl can serve the snRNA-seq arm without consuming excessive block face.
Same-day processing keeps paired sections synchronized. Cut the spatial slide and the snRNA-seq curl in the morning. Run the S200+ FFPE protocol (about 60 minutes total, less than 5 minutes hands-on) while the spatial slide processes on-instrument. Both datasets are initiated on the same day from the same block face.
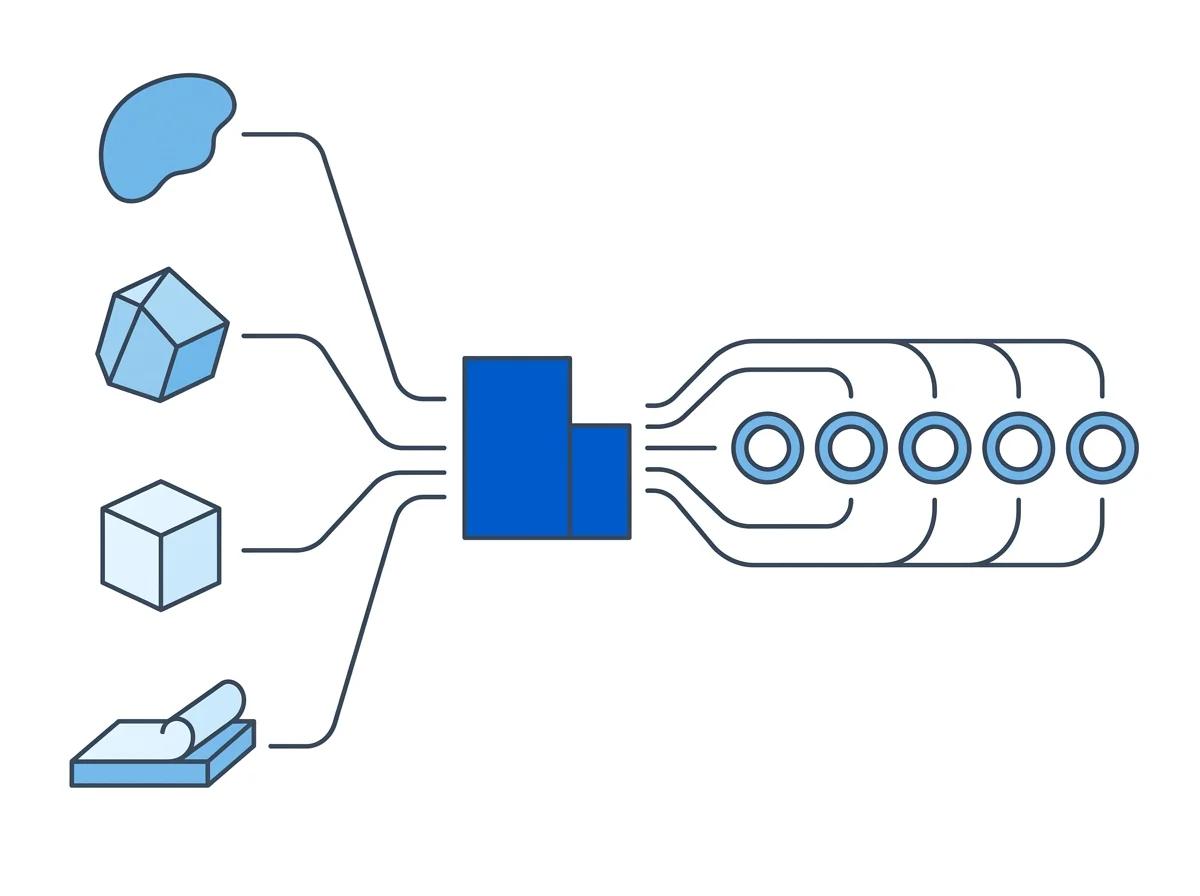
Match nuclei to platforms Match your nuclei to the right downstream platform
The Singulator 200+ produces nuclei that are not tied to any single downstream chemistry or vendor. This matters because multi-omic brain studies often span multiple analytical platforms, and the choice of sequencing or spatial technology should be driven by the scientific question -- not by sample prep constraints.
Validated platforms for S200+ brain FFPE nuclei
Choosing the right combination
For most brain FFPE studies, the default pairing is Xenium spatial + 10x Flex snRNA-seq. Xenium provides the spatial map; Flex provides the whole-transcriptome depth from dissociated nuclei. If your study focuses on rare cell populations -- disease-associated microglia in an Alzheimer's cohort, infiltrating T cells in a glioblastoma -- adding PERFF-seq to the analytical plan allows targeted capture of populations too rare for standard snRNA-seq to characterize at depth.
The S200+ two-cartridge workflow -- GREEN for deparaffinization followed by YELLOW NIC+ for nuclei isolation -- produces a clean nuclei suspension compatible with all of these platforms. No protocol modifications needed between Flex and PERFF-seq. The same nuclei prep feeds whichever platform your study requires.
Stanford and MSKCC validated PERFF-seq with Singulator 200+ nuclei for rare cell sequencing from FFPE tissue. Because PERFF-seq captures individual cells before library preparation, it recovers populations too sparse for droplet-based methods. For brain tissue, this means characterizing disease-specific populations that might represent less than 1% of all nuclei.
Feature Barcode technology (antibody-based protein detection) is not compatible with FFPE tissue on the Flex platform. Formalin fixation disrupts antibody-epitope interactions, making surface protein detection unreliable. For multi-omic protein + RNA analysis of brain tissue, use fresh-frozen sections with standard single-cell methods rather than FFPE.